Subspecies of the human gut microbiota carry implicit information for in-depth microbiome research
Abstract
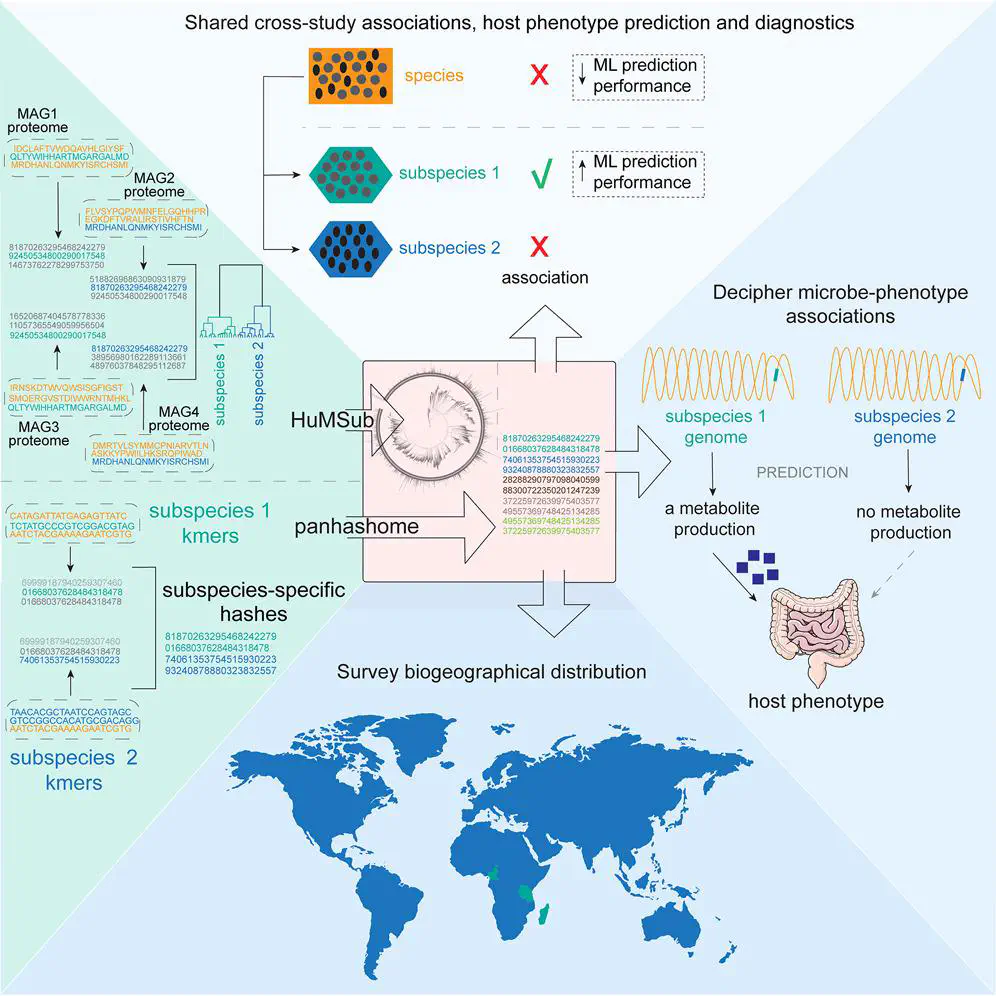
Microbial strains within a single species can exhibit distinct functional characteristics due to variations in gene content and often show individual specificity, which can obscure unbiased associations and hinder deductive research. Here, we comprehensively define the human gut microbiota at a consistently annotated operational subspecies unit (OSU) resolution in an unbiased, cohort-independent manner, demonstrating that this approach can generalize across diverse global populations while maintaining specificity and improving interstudy reproducibility. We develop panhashome—a sketching-based method for rapid subspecies and species quantification and identification of genes that drive intraspecies variations—and show that subspecies carry implicit information undetectable at the species level. We identify subspecies associated with colorectal cancer (CRC) whose sibling subspecies or species are not, while a machinelearning CRC diagnostic algorithm based on subspecies outperformed species-level methods. This subspecies catalog allows identification of genes that drive functional differences between subspecies as a fundamental step in mechanistically understanding microbiome-phenotype interactions.
Most microbiome studies stop at the species level. But strains within a species can differ dramatically in function. This obscures associations and limits reproducibility.
Going down to the strain-level isn’t useful for most comparative studies, because everyone has person-specific strains for most of their microbes!
We need an intermediate level: Subspecies.
We developed an unsupervised method to cluster genomes into operational subspecies units (OSUs) and built HuMSub: the most comprehensive catalog of human gut microbiota subspecies (5,361 OSUs across 977 species). We found that 1/3 of species in the human gut have subspecies, most of which we knew nothing about.
We created a sourmash-based method for fast & accurate subspecies quantification, enabling large-scale analysis across entire datasets, e.g., the whole SRA.
Why it matters: Subspecies explain disease associations invisible at the species level. For example, in colorectal cancer, subspecies-based ML models consistently outperformed species-level ones.
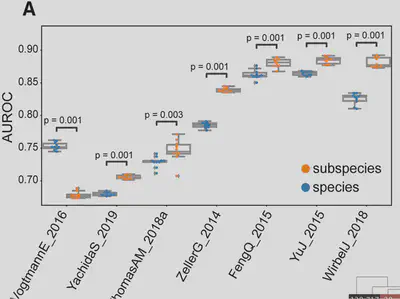
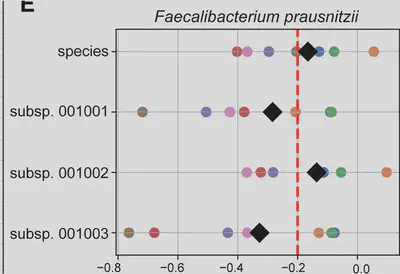
We found many cases where only one subspecies associates with disease while sibling-subspecies don’t. Sometimes no association is detected at species level because opposing subspecies associations cancel each other out.
Because our subspecies are genome-based, we can identify specific genes or mutations that explain differential associations. This provides a direct path to mechanistic understanding.