The easiest way to run jobs on a HPC cluster!
Why we need Snakemake?
Running Jobs on a HPC the old way
Imagine you have to run a job on a HPC cluster for many samples.
Here I illustrate it with running metaphlan, a typical tool for metagenomic analysis, but it could be any other tool.
Your HPC’s documentation tells you to write a bash script like this to submit it to a slurm cluster.
##!/bin/bash
#SBATCH --job-name=example_job
#SBATCH --output=example_job.out
#SBATCH --error=example_job.err
#SBATCH --time=01:00:00
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem=16G
metaphlan \
--input /path/to/input.fastq \
--output /path/to/output \
--threads 4 \
--memory 16G
Multiple samples
Now for multiple samples you can write a for loop, but then you do not benefit of the parallelization of the cluster. You can write a bash script that creates and submits the job scripts for each sample.
Something like this:
for sample in $(cat samples.txt); do
printf "#!/bin/bash
#SBATCH --job-name=${sample}_job
#SBATCH --output=${sample}_job.out
#SBATCH --error=${sample}_job.err
#SBATCH --time=10:00:00
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem=16G
metaphlan \\
--input /path/to/${sample}_1.fastq,/path/to/${sample}_2.fastq \\
--output \"/path/to/output/${sample}\" \\
--threads 4 \\
--memory 16G
" > /scripts/${sample}.sh
chmod +x /scripts/${sample}.sh
sbatch /scripts/${sample}.sh
done
Then you submit the script to the cluster and wait …
and get many error messages. Because:
Potential errors
- You missed a single backslash
- You forgot to create the output directory
- You did not correctly escaped a character
I speak of experience here!
Bash scripting
- Hard to read = Hard to debug
- One error screws up everything
- A lot of manual work
The new way: Pipeline languages 🥳
- Snakemake
- Nextflow
- CWL
I will focus on Snakemake.
Snakemake
In snakemake you would write the rule to run the metaphlan command like this.
rule run_metaphlan:
input:
R1="/path/to/{sample}_1.fastq",
R2="/path/to/{sample}_2.fastq"
output:
directory("/path/to/output/{sample}")
log:
"/path/to/{sample}.log"
threads: 4
resources:
mem_mb=16000
time_min=600
shell:
"metaphlan {input.R1},{input.R2} "
" --output {output} "
" --threads {threads} "
" --memory {resources.mem_mb} &> {log}"
Snakemake is a workflow management system that is written on top of Python. But as the example above shows, you mostly don’t need any python knowledge to write a snakemake pipeline.
Sample table
To define the input files for each sample you can identify them from the filenames in a predefined folder. What I use more is to define a sample table in a csv file with the paths to both fastq files for each sample.
import pandas as pd
SampleTable = pd.read_csv("samples.csv")
rule run_metaphlan:
input:
R1=SampleTable.loc[sample, "R1"],
R2=SampleTable.loc[sample, "R2"]
output:
directory("/path/to/{sample}")
...
Target rule
Then you need a target rule, that defines the final output of the pipeline.
This is analogous to the all target in a Makefile.
import pandas as pd
SampleTable = pd.read_csv("samples.csv")
rule all:
input:
expand("/path/to/output/{sample}", sample=SampleTable["sample"])
rule run_metaphlan:
input:
R1=SampleTable.loc[sample, "R1"],
R2=SampleTable.loc[sample, "R2"]
output:
directory("/path/to/output/{sample}")
...
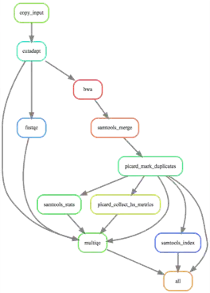
DAG
Writing what you need at the end might be sometimes a bit head-bending, but it is most of the time the results for all samples.
Snakemake defines a Directed Acyclic Graph (DAG) that defines which tool/job needs to be run for which input to get which output.
Obviously, you can also write a intermediate rule that uses the output from all samples and combines them.
here is the DAG for a real world example, where I run metaphlan on many samples and then combine the results into a single file.
Real world example

There is more!
Install Software automatically
Using conda or mamba you can install the software automatically in a conda environment.
So you can also install the pipeline on a new computer without having to install the software manually.
rule run_metaphlan:
...
conda:
"envs/metaphlan.yaml"
...
envs/metaphlan.yaml
channels:
- conda-forge
- bioconda
- defaults
dependencies:
- metaphlan =4.1
Run the pipeline with conda snakemake --use-conda
Singularity
You can even encapsulate each step in a singularity container to be sure that the software is run in a reproducible environment.
rule run_metaphlan:
...
singularity:
"docker://joseespinosa/docker-r-ggplot2"
...
Run the pipeline each step in a singularity container
snakemake --use-singularity.
Running on the cluster
Finally to submit the jobs to a slurm cluster you simply need to install the snakemake-executor-plugin-slurm, or another plugin for the cluster system you have and then you simply need to :
conda install -c bioconda snakemake-executor-plugin-slurm
snakemake --executor slurm --jobs 10
This command:
- Submits job to the cluster automatically
- Sets memory, threads, and time
- Tracks which job is done, or failed
- Saves logs
- Optimizes running time and disk usage
- Kills jobs if you stop the pipeline
- And resumes jobs if you restart the pipeline
Almost incredible!
Resources
- Snakemake Documentation
- Snakemake Tutorial
- I can help you with the rest.
Tips and Tricks
Create a profile for cluster usage
Note
This is the profile I use for running snakemake on the Ubelix cluster at the University of Bern. You can adapt it to your cluster system.
In ~/.config/snakemake/profiles/cluster create a file called config.yaml with the following content:
executor: slurm
jobs: 20 # max number of jobs to be submitted at once, can be higher.
## Default resources, if you do not define them in the rule
default-resources:
mem_mb: 1000
time_min: 120
rerun-incomplete: true # Recommended: if a job is halve done re-start it
use-conda: true # (as you prefer) use conda environments
conda-prefix: /path/to/conda/envs # where to store the conda environments.
# exceptions for specific rules
set-threads:
resource_intensive_rule: 50
set-resources:
big_mem_rule:
runtime: 5d
quos: job_cpu_long # here I set the slurm quos/pipeline
As the exceptions are mostly workflow specific, you can define a global profile for cluster usage and specific --workflow-profile options for each workflow.
Run Snakemake over a long time
I usually run snakemake in a screen session, as the main thread mostly submits jobs to the cluster and checks their status.
If you know that your pipeline works most of the time, you can add the flag --keep-going to the snakemake command. So that if there is an unexpected error the pipeline continues with other jobs.
Do not use this flag if you are developing the pipeline and you are not sure that a job will run successfully.